
本期,边肖将为您带来如何利用痛风石融合来鉴定融合基因的信息。文章内容丰富,从专业角度进行分析和描述。看完这篇文章,希望你能有所收获。
Tophat-fusion是一种利用RNA_seq数据识别融合基因的工具。
该软件集成在to fat软件中,只有在安装了to fat后才能使用。使用方法比较简单,唯一需要注意的就是目录结构。
Tophat-fusion需要一个固定的目录结构。例如,如果我在结果文件夹中分析tophat-fusion,那么我需要在这个目录中准备以下文件。
物种对应的RefGene.txt和ensGene.txt,可从UCSC下载。
创建一个新的爆炸文件夹。请注意,文件夹的名称必须是“blast”。在blast文件夹中,您需要从NCBI下载所有以NT、人类基因组和其他基因组开头的文件。下载链接如下:
FTP ://FTP . NCBI . NLM . NIH . gov/blast/db/
输出目录,每个样本对应一个输出目录。输出目录的前缀是tophat_,示例的名称添加在下划线之后,类似于MCF的tophat_MCF,其中MCF是示例的名称。
当然,你还需要对应物种的bowtie1的索引文件。请注意,它必须是bowtie1的索引。tophat在检测融合基因时推荐bowtie1的指数法。
上述所有文件准备就绪后,即可开始分析,步骤如下
00-1010的第一步实际上是使用tophat将读数与参考基因组进行比较。但是对于融合基因的读者来说,比较的方法比较特殊,需要增加额外的参数。具体代码如下
tophat 2-oto phat _ mcf 7-P20-fusion-search-keep-fasta-order-bow tie 1-no-coverage-search-r0-mate-STD-dev 80-max-intron-length 100000-fusion-min-dist 1000 00-fusion-anchor-length 13-fusion-ignore-chrmhg 19 _ bow tie 1/Hg 19 SRR 064286 _ 1。fastqsrr064286 _ 2。fastq
1. 比对参考基因组
在结果目录中,直接运行下面的代码就可以了。
top-fusion-post-P20-num-fusion-read S1-num-fusion-Pais 2-num-fusion-二者5hg19 _ bowtie1/hg19默认处理人类的融合基因。如果是其他物种,就要加上-非人参数。
Hat-fusion会根据目录结构自动识别对应的样本。操作完成后,会生成一个名为tophat-fusion _ out的文件夹,在该文件夹中会找到所有样本的融合基因分析结果。
我们只需要看看result.html文件,如下所示。
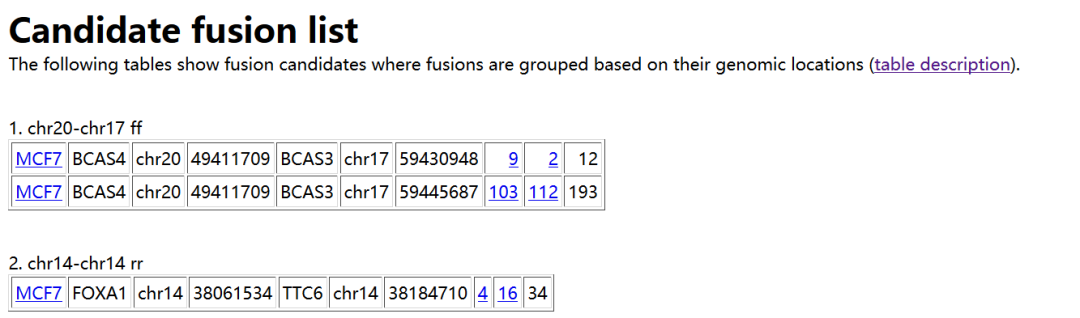

每一栏的含义如下
识别融合的样本名称
融合基因在“左边”
左边的染色体标识
左边的坐标
基因在“右边”
右边是染色体标识
右边的坐标
跨越读取次数
跨越配对的数量
一端跨越融合的跨越配对数
与fusionmap相比,这个软件的运行时间特别长。
这就是如何识别与上述边肖共有的痛风石融合的融合基因。如果你恰好也有类似的疑惑,可以参考上面的分析来理解。想了解更多,请关注行业信息渠道。
内容来源网络,如有侵权,联系删除,本文地址:https://www.230890.com/zhan/80653.html